Molecular Medicine
-
About
-
Molecular Medicine
Detecting Gene Alterations in Cancers
Molecular genetics laboratories employ multiple different methods to detect the variety of genetic alterations that are therapeutically relevant in cancers. For solid tumors, most of these tests are performed on formalin-fixed paraffin-embedded (FFPE) cancer tissues. Therefore, we review here practical techniques for detection from FFPE tissues.
The main types of tests fall into three major classes: those that analyze DNA, those that analyze mRNA expression, and those that analyze protein expression. Results from DNA, mRNA, and protein analyses can have different implications. Furthermore, although all three classes of tests can analyze the same target in a tissue, the clinical significance can vary. For example, in lung cancer, EGFR DNA mutations predict very well sensitivity to EGFR tyrosine kinase inhibitors, while EGFR DNA copy number levels, mRNA expression levels, or protein levels have little or no predictive value.
DNA-based alterations
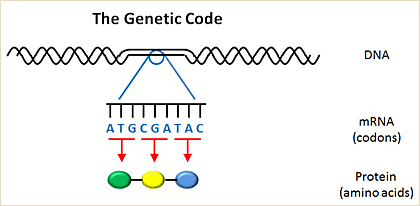
Genetic code
Genetic information is passed from one cell to another in the form of deoxyribonucleic acid (DNA). DNA encodes messenger ribonucleic acid (mRNA), which is then transcribed into protein. The building blocks of DNA involve four nucleotides, including adenosine (A), cytosine (C), guanine (G), and thymidine (T). The building blocks of proteins are amino acids, of which there are 20. The human genome is comprised of about 3 billion nucleotides. Of these 3 billion nucleotides, only ~5% encodes for genes which are translated into proteins within a cell. A gene is further divided into exons, which contain the actual information used in coding for protein synthesis, and introns, which are segments between exons that are removed before the protein is translated (introns do not contain any coding information).
Nucleotides are organized into three letter code words called codons. Each codon encodes for a single amino acid. The genetic code is the entire set of 64 three letter codes that convert codons into amino acids. Notably, the genetic code is degenerate; in other words, because there are 64 codons and only 20 amino acids, more than one codon can encode for the same amino acid. Some codons are called stop codons, because instead of encoding an amino acid they tell the cell which amino acid is the last one in a protein. Mutations alter the normal sequence pattern of DNA. Somatic mutations arise in cancers but are not found in matched normal tissues from the same patient.
Point mutations
Point mutations (such as EGFR L858R in lung cancer) result from single nucleotide substitutions. If these mutations occur within in exons, they can either be synonymous (i.e., the encoded amino acid stays the same) or non-synonymous (i.e., the encoded amino acid changes). For detection of recurrent point mutations, direct dideoxynucleotide (Sanger) sequencing has historically been the gold standard. However, this method usually involves analysis of one gene at a time and has low sensitivity, most often requiring more than 25% tumor cells in a given specimen.
More recently, assays have been developed to detect tens of mutations concurrently, using either mass spectrometry or capillary electrophoresis (Dias-Santagata et al. 2010; Fumagalli et al. 2010; Lurkin et al. 2010; Su et al. 2011). Both assays rely upon multiple PCR (polymerase chain reaction) steps to amplify DNA regions of interest and multiplex single-base primer extension with dideoxynucleotides. Extension products are then analyzed either by mass spectrometry, which can distinguish different bases according to their mass-to-charge (m/z) ratio (Sequenom Mass Spectrometry system) or by capillary electrophoresis, which distinguishes bases by size and by the color of fluorescently labeled nucleotides (Applied Biosystems SNaPshot system). Both methods are more sensitive than direct sequencing and can be performed on small quantities of FFPE-derived tumor DNA.
Insertions/deletions
Insertions (such as HER2 exon 20 insertions in lung cancer) and deletions (such as EGFR exon 19 deletions in lung cancer) result when nucleotides are inserted or deleted, respectively, in coding portions (exons) of the genome. Insertions and deletions can be detected by direct sequencing of the exons of interest. An alternative method involves PCR-based sizing assays, in which fluorescently labeled primers are used to amplify a targeted DNA sequence across the region of interest. PCR products are then size-separated very accurately by capillary electrophoresis, allowing for easy detection of small insertions or deletions (causing small size shifts). Although sizing assays do not reveal the exact sequence of the insertions/deletions, they are more sensitive than direct sequencing (Pan, Pao, and Ladanyi 2005). They are also more comprehensive and can detect any insertion/deletion, without the need for allele-specific primers which could miss detecting certain mutations.
Gene amplification/fusions
Regions of DNA that encode genes can become amplified; in other words, instead of having the normal 2 copies (one from each parent), cells acquire more copies (such as HER2 gene amplification in breast cancer). Regions of DNA can also become abnormally rearranged; in other words, regions of DNA not normally next to each other become fused together (such as fusions of the EML4 and ALK genes in lung cancer).
Gene amplification and/or rearrangements are commonly detected by the method of fluorescence in situ hybridization (Penault-Llorca et al. 2009; Cataldo et al. 1999). DNA probes targeting specific regions are fluorescently labeled and hybridized to tissue specimens. Normal cells should have 2 copies of the target gene; multiple copies suggest gene amplification. In the case of gene fusions, a dual probe 'break-apart' FISH assay is typically used, where 2 probes labeled with different fluorescent "colors" that hybridize on either side of the gene of interest are utilized. If the gene is intact, the 2 probes are so close that they overlap and the colors merge together, but if the gene is rearranged, the 2 probes become abnormally separated. The use of break-apart probes can detect if a rearrangement is present, but a limitation is that the 'partner' gene fused with the rearranged gene will not be known. Other methods (such as RT-PCR (reverse transcriptase polymerase chain reaction)) are needed to identify the specific gene fusion (Choi et al. 2008).
Gene deletion/non-recurring mutations
In contrast to oncogenes, which are typically activated by specific, recurrent events such as gene fusion, gene amplification, or mutations in a few, critical positions ("hotspot mutations"), the inactivation of tumor suppressor genes occurs through a much more heterogeneous spectrum of genetic alterations, for which testing is therefore far more laborious and complicated. Because of this, clinical testing of archived tumor material for tumor suppressor gene alterations is still not routinely or widely performed. Immunohistochemistry with protein-specific antibodies can be used to detect lack of protein expression. Direct sequencing can be used to seek mutations across an entire gene.
mRNA-based alterations
mRNAs can either be present or absent in cancers. When present, they can sometimes be 'overexpressed' (expressed at higher levels than normal). mRNA levels are detected in tumor tissue using a process called quantitative RT-PCR (reverse transcriptase polymerase chain reaction). Mutations can also be detected using mRNA. Generally, since mRNAs are much less stable compared to DNA, they are often too degraded for analysis in clinical samples; thus, it is easier to use DNA to detect mutations. One exception to this is that the exact sequence of known gene fusions is more readily detected at the mRNA level.
Protein-based alterations
Immunohistochemistry
Proteins can either be present or absent in cancers. When present, they can sometimes be 'overexpressed' (expressed at higher levels than normal). Proteins are detected using a process called immunohistochemistry (IHC). IHC involves 'staining' tissues with antibodies that recognize specific proteins of interest. As long as such specific antibodies are available and reliable, IHC can be used to detect mutated proteins or the lack thereof in tumor tissue. However, protein levels may or may not be clinically relevant. For example, in lung cancer, EGFR DNA mutations but not EGFR protein levels by IHC predict for sensitivity to EGFR tyrosine kinase inhibitors. However, a recent study (Pirker et al. 2012) suggests that high levels of EGFR expression, as measured using IHC, may predict response to cetuximab (an anti-EGFR antibody) plus chemotherapy as compared to chemotherapy alone.
Suggested Citation: Pao, W., M. Ladanyi. 2015. Detecting Gene Alterations in Cancers. My Cancer Genome https://www.mycancergenome.org/content/molecular-medicine/detecting-gene-alterations-in-cancers/ (Updated February 17).
Last updated: May 26, 2019
Disclaimer: The information presented at MyCancerGenome.org is compiled from sources believed to be reliable. Extensive efforts have been made to make this information as accurate and as up-to-date as possible. However, the accuracy and completeness of this information cannot be guaranteed. Despite our best efforts, this information may contain typographical errors and omissions. The contents are to be used only as a guide, and health care providers should employ sound clinical judgment in interpreting this information for individual patient care.
