Molecular Medicine
-
About
-
Molecular Medicine
Types of Molecular Tumor Testing
Alterations in genes encoding cellular signaling molecules, especially protein kinases, can result in cancers. Drugs targeting mutant kinases are efficacious in cancer patients. The sensitivity of such drugs is related to the genetic makeup of individual tumors. Thus, mutational profiles of tumor DNA help prioritize anti-cancer therapy and direct patient management.
Many types of gene alterations can occur in cancers. The four main types include:
- Single nucleotide variants (SNVs), also known as point mutations. SNVs result from a base substitution at one nucleotide. These may result in a change in the amino acid sequence of the encoded protein (missense mutation) or a premature truncation of the protein (nonsense mutation).
- Small duplications of consecutive nucleotides, insertions or deletions involving one or a few nucleotides, or more complex mutations involving simultaneous deletions and insertions of one or a few bases (indelsa). These types of mutations may be “in-frame,” resulting in the addition or subtraction of amino acids in the protein, or can cause a “frameshift,” typically resulting in a premature truncation of the protein.
- Exon or gene copy number changes. Exon copy number changes include large duplications or deletions encompassing entire exons and affecting the functional domains of the protein. Gene copy number changes include amplifications or deletions of the entire gene.
- Structural variants (SVs) or large structural anomalies of genetic material including translocations or inversions that result from breakpoints between multiple chromosomes or within a single chromosome. These often result in fusion genes and associated fusion proteins.
Often, cancer-causing mutations cluster in “hotspots” where tumors from different patients harbor the same recurrent mutation. Some hotspot SNVs may occur frequently, while others are rare. For example, the BRAF V600E mutation occurs in 40% of all melanomas, while the BRAF L597S mutation occurs in <1% of all melanomas.
Tests for molecular profiling of tumors range from simple to complex. The simplest tests detect only one type of mutation in one gene, such as an SNV. An example is a test that looks for only the specific T to A substitution mutationat position c.1799 of BRAF, resulting in the amino acid substitution of glutamic acid for valine (p.V600E) in the protein. The V600E mutation is commonly observed in melanoma. Conversely, the most complex tests can simultaneously detect all the major types of gene alterations, including substitutions, duplications, insertions, deletions, indels, gene copy number variations, and structural variants, including inversions and translocations. An example is a test offered by Foundation Medicine, called FoundationOneTM, which tests for mutations in about 322 genes commonly altered in cancer. Whole genome sequencing (WGS) will also detect the four main types of gene alterations, providing a comprehensive analysis of the 20,000+ genes in the human genome.
Clinical testing for most genetic variants is performed in a CLIA-certified molecular genetics or molecular pathology laboratory using technologies such as allele-specific polymerase chain reaction (PCR), Sanger dideoxy sequencing, pyrosequencing, multiplex ligation-dependent probe amplification (MLPA), or mass spectrometry (MS). Clinical testing for gene copy number and structural variants is usually performed in a separate CLIA-certified cytogenetics laboratory using fluorescence in situ hybridization (FISH).
Newer, next-generation sequencing (NGS) technologies, such as massively parallel sequencing, will change the way laboratory tumor molecular profiling is performed. NGS technologies allow for whole exome sequencing, which examines all protein-coding regions in the human genome, or whole genome sequencing, which analyzes both protein-coding and non-coding regions in the human genome. Examples of machines used in NGS include the Illumina MiSeq and HiSeq and the Life Technologies Ion Torrent PGM and Proton.
Those interested in molecular tumor testing should understand the wide variety of testing platforms available and the sensitivities and specificities of each. Many clinical laboratory tests use laboratory-developed assays, while some commercial kits offer gene-specific mutation testing. Prior to patient testing, all assays in a CLIA-certified laboratory must be validated. Additionally, all laboratories are required to perform semiannual proficiency testing by genotyping unknown, blinded proficiency samples to ensure the accuracy of their assays and interpretation of their results. Tumor molecular profiling is generally performed on DNA extracted from formalin-fixed paraffin-embedded tissue specimens. Prior to testing, the tissue specimen requires examination by a pathologist to confirm the presence of the tumor and to determine the tumor content in the specimen. Table 1 summarizes testing modalities and the variants they detect. Following the table are further details about each testing modality.
| Table 1. Types of Molecular Tests and Variants Detected. | ||||
| Molecular Methodology | Variant Types | |||
|---|---|---|---|---|
| SNVs | Small duplications, insertions, deletions, indels | Exon duplications, deletions or gene copy number changes | SVs | |
| Allele-specific PCR | ✓ | |||
| PCR and Sanger dideoxy sequencing | ✓ | ✓ | a | |
| PCR and pyrosequencing | ✓ | • | ||
| PCR and MS | ✓ | • | ||
| PCR and single base extension | ✓ | |||
| MLPA | ✓ | ✓ | ||
| FISH | b | ✓ | ||
| NGS – custom panels (amplicon capture) | ✓ | ✓ | ||
| NGS – custom panels (hybridization capture) | ✓ | ✓ | ✓ | c |
| NGS – whole exome sequencing | ✓ | ✓ | ✓ | • |
| NGS – whole genome sequencing | ✓ | ✓ | ✓ | ✓ |
| ddPCR | ✓ | ✓ | c | |
| BEAMing | ✓ | ✓ | c | |
| NOTES: ✓ Variant detected. • Variant detected with difficulty. a Variant detected if fusion RNA is extracted first. b Only gene copy number variants detected. c Structural variants resulting from well-known, frequently occurring breakpoints are relatively straightforward to include in this type of assay, while structural variants resulting from a wider variety of possible breakpoints are less likely to be included or, therefore, detected. See descriptions below for further detail. FISH = fluorescence in situ hybridization; indels = mutations including both insertions and deletions; MLPA= multiplex ligation-dependent probe amplification; MS = mass spectrometry; NGS = next-generation sequencing; PCR = polymerase chain reaction; SNVs = single nucleotide variants; SVs = structural variants. |
||||
Allele-Specific PCR
Variants detected: Typically considered targeted analysis for the detection of a specific SNV.
Method: Real-time PCR with fluorescent reporter probes in which reporter probes for one wild type and one mutant are added to the reaction mixture. Following hybridization to the genomic DNA, polymerase extends the probes in a complementary fashion, releasing the reporter molecules for detection. Subsequent PCR cycles using the reporter probes result in amplified signals, allowing for precise measurement of one or both alleles of interest. Alternatively, routine fluorescent PCR coupled with capillary electrophoresis for detection is used. Similarly, the 3’ end of the mutant-specific primer is extended only in the presence of DNA with that mutation.
Time to complete test: 1–2 days.
Pros: Sensitive—can detect mutant DNA if present at 1–5%. No special equipment required.
Cons: Target specific and cannot detect other mutations that may be present in tumor DNA.
Sanger Dideoxy Sequencing
Variants detected: Unknown mutations including SNVs and small duplications, insertions, deletions, and indels of interest.
Method: Sanger dideoxy sequencing is typically performed on PCR products. Sequencing primers hybridized to the PCR product and are extended using DNA polymerase, the four deoxynucleotides (dNTPs), and a mixture of fluorescently labeled dideoxynucleotides (ddNTPs). Each of the four ddNTPs is labeled with a different fluorescent dye. Random incorporation of the labeled ddNTPs results in termination of strands at each location along the sequence. The reaction primers can be extended by up to about 1,000 nucleotides. Capillary electrophoresis separates the strands by size, and the terminating nucleotides are measured using fluorescence spectroscopy. In a clinical laboratory, both the forward and reverse strands are sequenced.
Time to complete test: 2–3 days.
Pros: A variety of unknown mutations can be detected. Can be used to detect gene fusions if RNA from the fusion transcript is first extracted from the specimen. No special equipment required.
Cons: Labor intensive and requires mutant DNA to be present at 20–25%. Cannot detect changes in exon or gene copy number.
Pyrosequencing
Variants detected: Unknown mutations in a small targeted region.
Method: A sequencing primer is hybridized to the PCR product and is extended with deoxynucleotides in a complementary fashion in the presence of ATP sulfurylase, luciferase, apyrase, adenosine 5’-phosphosulfate, and luciferin. Pyrophosphate is generated when a base is incorporated, and through a series of reactions, light is generated. The sequence can be determined from the order of the dNTP additions and the intensity of the light produced with each addition.
Time to complete test: 2–3 days.
Pros: Quick and sensitive detection of mutant DNA at a level of 5%.
Cons: Requires pyrosequencing instrumentation; is limited in the types of mutations that can be detected in tumor DNA.
Mass Spectrometry – MS
Variants detected: Targeted SNVs.
Method: Matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS; e.g., Sequenom). Detects 1–100+ SNVs simultaneously. These tests are custom-designed to interrogate tissues for mutations of interest and are usually designed to look for common mutations that occur at specific hotspots. First, primer extension is performed using site-specific primers and a combination of dNTPs and ddNTPs, selected such that different alleles will result in products of different (predictable) sizes. Primer extension products are resolved by mass spectrometry in order to determine the genotype.
Time to complete test: 2–3 days.
Pros: Sensitive, reliably detecting mutant DNA if present at 5–10%; tests more than one gene.
Cons: Requires mass spectrometry instrumentation; SNV-specific and cannot detect other mutations in tumor DNA that may be present.
Single Base Extension Assay
Variants detected: Targeted SNVs.
Method: Multiplex PCR, multiplex primer base extension coupled with capillary electrophoresis (e.g., Life Technologies' SNaPshot® Multiplex System). Detects 1–100+ SNVs simultaneously. These tests are custom-designed to interrogate tissues for mutations of interest and are usually designed to look for common mutations that occur at specific hotspots.
Time to complete test: 2–3 days.
Pros: Sensitive, reliably detecting mutant DNA if present at 5–10%; tests more than one gene. No special equipment required.
Cons: SNV-specific and cannot detect other mutations that may be present in tumor DNA.
Multiplex Ligation-Dependent Probe Amplification – MLPA
Variants detected: Exon and gene copy number. Depending on experimental design, can also detect SNVs.
Method: A series of steps that begins with specially designed primers that hybridize to patient DNA. Following hybridization, the bound primers are enzymatically ligated and the ligation products are amplified using multiplex PCR. Amplified products are detected and analyzed by capillary electrophoresis.
Time to complete test: 2–3 days.
Pros: Quick and able to detect multiple mutations simultaneously. No special equipment required. Can detect targeted SNVs at 10%.
Cons: For exon or gene copy number variant detection, requires mutant DNA to be present at levels of 20–40%. Both targeted SNV and exon and gene copy number variants are specific, and other mutations in tumor DNA cannot be detected. Kits may not be available for the gene or mutation of interest. Testing works better on fresh frozen tissue than on DNA extracted from paraffin-embedded tissue.
Fluorescence In Situ Hybridization – FISH
Variants detected: Targeted gene copy number changes and targeted SVs.
Method: Fluorescent probes are used to locate genes or sequences of interest on one or more chromosome. Fluorescence microscopy is used for detection.
Time to complete test: 2–3 days.
Pros: Easily detects gene copy number changes and targeted SVs that are not as easily detected by other methods; cell-based imaging enables detection of events in a small fraction of cells.
Cons: Requires paraffin-embedded tissue on unstained slides; cannot detect most types of mutations occurring in solid tumor neoplasms.
Next-Generation Sequencing – Custom Panels, Amplicon Capture
Variants detected: Substitutions, duplications, insertions, deletions, and indels.
Method: Multiplexed PCR amplification of regions of interest, followed by NGS and specialized bioinformatics analysis. Examples of amplicon capture technology include Ion Torrent AmpliSeq, Illumina TruSeq, Agilent HaloPlex, and RainDance. These tests are custom-designed to interrogate tissues for mutations of interest in specific genes, typically 1 to 100.
Time to complete test: Several days to weeks, depending on complexity of the assay.
Pros: Enables simultaneous detection of single base substitutions as well as more complex mutations including duplications, insertions, deletions, and indels in many genes in a single assay; requires low inputs of DNA. When sequencing to high “depth of coverage” (1000x coverage), assays are sensitive to detect low abundance mutations. Compared to other NGS methods, these panels can be cheaper or faster, since they are more limited in scope.
Cons: Expensive; requires a completely different DNA preparation method than what is used for other molecular detection methods. Unable to detect gene copy number changes and SVs. Certain NGS platforms exhibit platform-specific artifacts, such as false positive insertion, deletion, and indel calls. Also, if the bioinformatics analyses are not optimized, these variants can also be missed. If 1000x coverage is used, the level of sensitivity can be so high that the significance of low-level mutations is unknown.
Next-Generation Sequencing – Custom Panels, Hybridization Capture
Variants detected: Substitutions, duplications, insertions, deletions, indels, exon and gene copy number changes, and select translocations.
Method: Hybridization capture of regions of interest, followed by NGS and specialized bioinformatics analysis. Genomic DNA fragments are hybridized in solution to sequence-specific capture probes corresponding to target regions of the genome. Examples of hybridization capture technology include Agilent SureSelect, Nimblegen SeqCap, and Illumina TruSeq. These tests are custom-designed to interrogate tissues for mutations of interest in specific genes, typically 50 to several thousand.
Time to complete test: Several days to weeks, depending on complexity of the assay.
Pros: Enables simultaneous detection of substitutions, duplications, insertions, deletions, indels, and exon and gene copy number changes in many genes in a single assay. Probes can also be designed to capture select translocation breakpoints in recurrently rearranged genes, as in FoundationOneTM. When sequencing to high depth of coverage (500–1000x coverage), assays are sensitive to detect low abundance mutations. Compared to other NGS methods, these panels can be cheaper or faster, since they are more limited in scope.
Cons: Expensive; requires a completely different DNA preparation method than has been used traditionally for other molecular mutation assays; requires more tumor tissue; requires sophisticated bioinformatics. Certain NGS platforms exhibit platform-specific artifacts, such as false positive insertion, deletion, and indel calls. Also, if the bioinformatics analyses are not optimized, these mutations can be missed. Depending upon the depth of coverage used, the assay can be insensitive (30x coverage) or extremely sensitive (1000x coverage). If 1000x coverage is used, the level of sensitivity can be so high that the significance of low-level mutations is unknown.
Next-Generation Sequencing – Whole Exome Sequencing
Variants detected: Substitutions, duplications, insertions, deletions, indels, and gene copy number changes.
Method: Hybridization capture of regions of interest, followed by NGS and specialized bioinformatics analysis. Genomic DNA fragments are hybridized in solution to sequence-specific capture probes corresponding to all protein-coding exons throughout the genome. These tests interrogate whole exomes.
Time to complete test: Several days to weeks.
Pros: Intermediate in comprehensiveness. Enables simultaneous detection of substitutions, duplications, insertions, deletions, indels, and exon and gene copy number changes in many genes in a single assay.
Cons: Expensive; requires a completely different DNA preparation method than has been used traditionally for other molecular mutation detection techniques; requires more tumor tissue; requires sophisticated bioinformatics. Certain NGS platforms exhibit platform-specific artifacts, such as false positive insertion, deletion, and indel calls. Also, if the bioinformatics analyses are not optimized, indels can be missed. Compared to custom panels, whole exome sequence typically produces lower depth of coverage, limiting the detection sensitivity for low-abundance mutations. The clinical significance of most genomic alterations detected by whole exome sequencing is unknown. Non-tumor specific, germline variants associated with disease will also be detected. Ethical concerns exist regarding the identification of these “incidental findings” are a consideration.
Next-Generation Sequencing – Whole Genome Sequencing
Variants detected: Substitutions, duplications, insertions, deletions, indels, gene copy number changes, and chromosome inversions and translocations.
Method: Random shearing of genomic DNA molecules, followed by NGS and specialized bioinformatics analysis. These tests interrogate both coding and non-coding regions of the human genome.
Time to complete test: Several days to weeks, depending on the NGS platform and analysis methods.
Pros: Most comprehensive. Enables simultaneous detection of substitutions, duplications, insertions, deletions, indels, gene and exon copy number changes, and chromosome inversions and translocations across the entire genome.
Cons: Expensive and low throughput; requires a completely different DNA preparation method than has been used traditionally for most mutation detection techniques; requires more tumor tissue; requires sophisticated bioinformatics; large computational demands for data storage and processing. Certain NGS platforms exhibit platform-specific artifacts, such as false positive insertion, deletion, and indel calls. Also, if the bioinformatics analyses are not optimized, indels can be missed. Compared to exon capture methods, whole genome sequencing typically produces lower depth of coverage, limiting the detection sensitivity for low-abundance mutations. The clinical significance of most genomic alterations detected by whole genome sequencing is unknown. Non-tumor specific, germline variants associated with disease will also be detected. Ethical concerns exist regarding the identification of these “incidental findings” are a consideration.
Digital Droplet PCR – ddPCR
Variants detected: Targeted known mutations, including SNVs, small insertions, deletions, and indels, and rearrangements.
Method: ddPCR is used to detect mutations in circulating tumor DNA (ctDNA) with high sensitivity and specificity. Circulating cell free DNA (cfDNA), which includes DNA derived from tumors (ctDNA), is isolated from the blood plasma of cancer patients. Extracted DNA is mixed with PCR reaction components, primers for genes of interest, and fluorescent hydrolysis probes specific for mutation or wild-type sequences. This aqueous mixture is emulsified into tiny droplets such that, on average, there is less than one haploid genome equivalent per droplet and used for emulsion PCR (ePCR) (Pekin et al. 2011). During ePCR , the fluorescent probes hybridize to amplified mutant or wild-type sequences present in the droplet and are cleaved during amplification to release the fluorophores. The emulsion is then reinjected into a microfluidic device which spaces the droplets, allowing for fluorescence detection for each individual droplet (Didelot et al. 2013; Hindson et al. 2011; Manoj 2016; Pekin et al. 2011; Pinheiro et al. 2012; Taly et al. 2012; Taly et al. 2013; Zhong et al. 2011). The recent development of multiplex readouts, which rely on both fluorescence color and intensity, has allowed for the simultaneous detection of ≥5 targets. ddPCR can include probes of one color, but at differing concentrations, such that the fluorescence intensity readout for same-colored probes to different target sequences varies with the choice of probe concentration (higher concentrations of probe results in a linearly brighter signal; Figure 1). Alternatively, multiple probe colors can be utilized at different concentrations and probe colors can be mixed for a single target. This results in a multiplex assay readout that capitalizes on both fluorescence color and intensity; in this way, the number of possible reactions increases geometrically with the addition of new probe colors, rather than linearly (Didelot et al. 2013; Zhong et al. 2011; Figure 2).
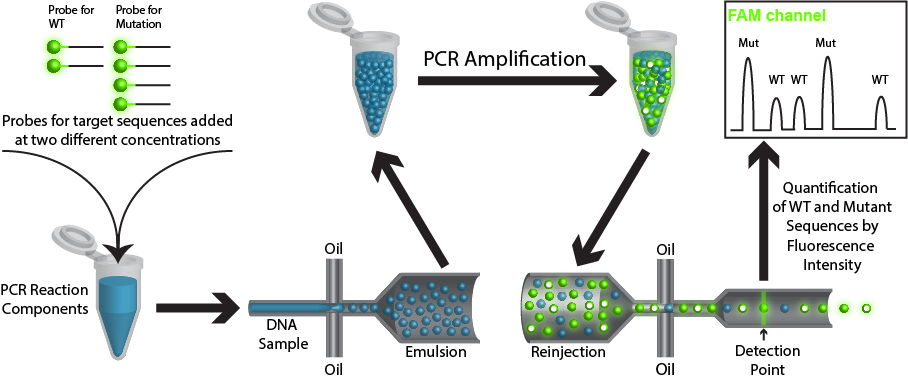
Figure 1. Simplified rendition of ddPCR showing the use of a single color probe at multiple concentrations. In this case, the probe for the wild-type (WT) sequence is added at half the concentration as the probe for the mutated (Mut) sequence. The sample is then emulsified such that each droplet contains all PCR reagents, probes, and ≤1 copy of template DNA. During PCR hydrolysis of the probes occurs to release the fluorophores. The post PCR sample is then reinjected and fluorescence on individual droplets is measured. Droplets that contain a WT sequence template will fluoresce at half the intensity as droplets containing a mutated template.
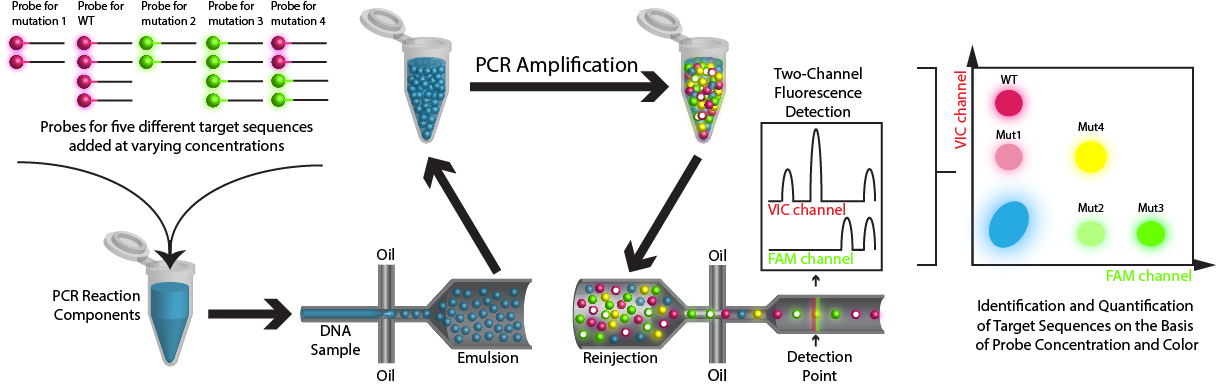
Figure 2. Cartoon rendition of ddPCR demonstrating that a multiplex readout is possible with the use of two probe colors and varying probe concentrations.
Time to complete test: 2–3 days.
Pros:: High level of sensitivity and specificity; relatively inexpensive.
Cons: Can only detect known targeted mutations; limited in the types of mutations detected; can only detect a limited number of mutations per assay.
Beads, Emulsification, Amplification, and Magnetics – BEAMing
Variants detected: Targeted known mutations, including SNVs, small insertions, deletions, and indels, and rearrangements.
Method: BEAMing is used to detect mutations in circulating tumor DNA (ctDNA). Circulating cell free DNA (cfDNA), containing ctDNA, is extracted from the blood plasma of cancer patients. The gene of interest is pre-amplified using PCR with primers specific for the gene of interest attached to tag sequences. The amplified DNA is mixed with beads coated with primers to capture the tag sequences and ePCR is performed. After ePCR, each emulsion droplet will contain a bead that is coated with thousands of copies of the original single DNA molecule. Next, fluorescent probes to mutations of interest are hybridized to the amplified DNA. Magnetic flow recovery methods can be used for detection. The detection limit of this assay is as low as one mutant copy among 10,000 DNA molecules ( Azvolinsky 2012; Diehl et al. 2006; Richardson and Iglehart 2012; Figure 1).

Figure 1. Cartoon depiction of the BEAMing method of ctDNA detection.
Time to complete test: 2–3 days.
Pros: High level of sensitivity and specificity
Cons: Can only detect known targeted mutations; limited in the types of mutations detected; can only detect a limited number of mutations per assay.
NOTES: aWhile the term indel is commonly used to refer to insertion, deletion, and combined insertion/deletion mutations, the Human Genome Variation Society (HGVS), viewed as the authority on genome variation nomenclature, defines indels as "deletion/insertions of two or more consecutive nucleotides" (see http://www.hgvs.org/mutnomen/disc.html#indel). This article follows HGVS convention.
Suggested Citation: Vnencak-Jones, C., M. Berger, W. Pao. 2016. Types of Molecular Tumor Testing. My Cancer Genome https://www.mycancergenome.org/content/molecular-medicine/types-of-molecular-tumor-testing/ (Updated February 8).
Last updated: Feb 03, 2020
Disclaimer: The information presented at MyCancerGenome.org is compiled from sources believed to be reliable. Extensive efforts have been made to make this information as accurate and as up-to-date as possible. However, the accuracy and completeness of this information cannot be guaranteed. Despite our best efforts, this information may contain typographical errors and omissions. The contents are to be used only as a guide, and health care providers should employ sound clinical judgment in interpreting this information for individual patient care.
